Understanding the landscape of histone readers is of crucial importance to my reserach. These proteins which recognize, and recruit various complexes and proteins to post tranlational modification (PTMs) cause many of the correlated effects of we recognize in epigenomic research. For example, when methylation is added to a lysine on the alpha globulin tail of a histone (K36 in this instance), this methylation has no direct effect on altering the chromatin like acetylation does. Rather it affects transcription by recruiting a series of readers which in turn recruit more factors. And so the chromatin cylcle of influcence continues.
An essential mark I use to identify novel transcription regions in monocots is H3K36me3. So in turn, I wanted to read a paper getting into the identification of a reader enzyme for this specific mark. Enter Jin et al, 2015, “MORF-RELATED GENE702, a Reader Protein of Trimethylated Histone H3 Lysine 4 and Histone H3 Lysine 36, Is Involved in Brassinosteroid-Regulated Growth and Flowering Time Control in Rice”. A paper diving into this very question.
Background
The basic set-up of this paper is this group had previously identifed two proteins in Arabidopsis that recognize H3K4me3 and H3K36me3, namely MORF RELATED GENE (MRG) 1 & 2 via their chromodomains, and a series of functional studies. Therefor, this group was curious how MRG would operate in monocots since it had only been tested in dicots. So, they chose to investigate this proteins function in rice.
Experiments
This group initially tested the expression of MRG702 and MRG 701, ortholgs of the Arabidposis version. Found that MRG 702 was the more up-regulated version, and used this as evidence to call it the more important functional copy.
They they generated knock-down version of 702, and both 701 and 702. They discovered that the knockdown of 702 had an almost identical phenotype to knocking out both, allowing the scientist to conclude that 702 is the more important protein here as the phenotype wasn’t exacterbated at all.
I have to say, the knockdown phenotypes look pretty good! They also had a smart decision here where they also knocked down the histone methyltransferase responsible for laying down H3K36me3. This allowed the authors to compare knockouts putativly in similar pathways. Interestingly similar phenotypes were found, pointing to the fact that these are more than likely operating in the same genetic pathway.

Figures #2 and #3 basically get at the phenotypes of these knockouts, and while they look somewhat striking, they rely on a few test of Brassinosteroid sensing, and flowering time that I don’t like very much. For first their assays, while I’m sure valuable, are incredibly domain specific, and make me question how valuable they really are.
Second, I have an issue with this studies repeated use of RT-PCR at a few select loci. When measuring complex phenotypes, they attempt to use previous literature to select loci that they beleive should have signifigantly different expression from their knockouts. While this appears to be true, I would have preffered if this lab had done RNA seq to look at a more global level of transcription. Since this reader is more than likely not targeted to this single pathway, I would have found it more convincing if they had demonstrated this on a global level.
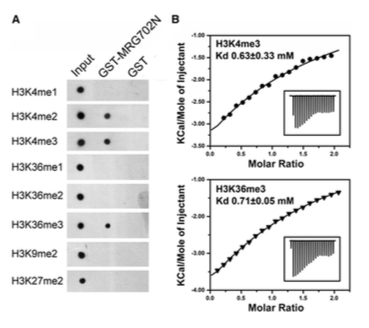
Figure #4 is in my opinion the second key figure of this entire paper. It illustrates that in a dot blot assay link: https://en.wikipedia.org/wiki/Dot_blot (a varient of western where you basically don’t get size information), you find specific binding of MRG702 for H3K36me3, and both H3K4me2 and H3K4me3. Basically, big poing MRG702 has histone specificity!!! Woo!
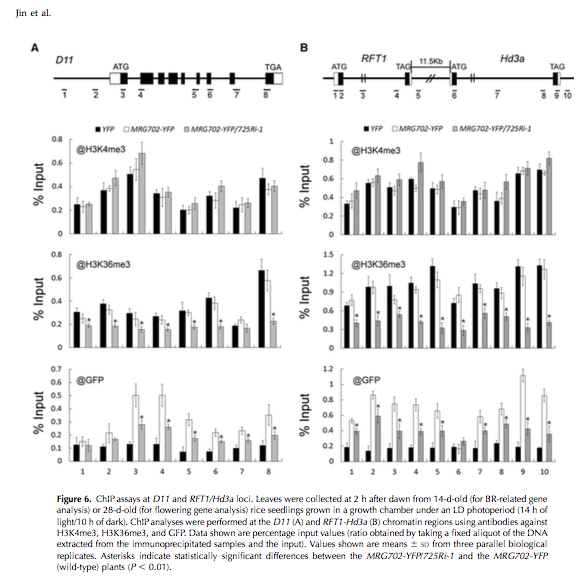
Finally, the authors do ChIP, and demonstrate that MRG702 binding acts on H3K36me3, and use a 725Ri-i mutant (SDG8 mutant) to demonstrate the MRG702 binds to known loci whith H3K36me3 deposited, and that in the abscence of this methylation (mutatnt), you no longer MRG binding. Pretty neat!!!
Final Thoughts
A solid paper overall that I believe did a decent job identifying a reader of H3K36me3 in Oryza sativa. I still wish the authors didn’t use rt-PCRl, and instead had done RNA-seq as well as ChIP-seq. But I recognize I have some intense genomic bias in this.