There’s a ton of papers out there. Some are good, some are bad, and some are really - really important. So, here is a collection of papers that I think are important, and one or two reasons why I think so. This is going to be broken down loosely by subject matter, and will be updated as I go along.
Enhancers
Genetic dissection of the alpha-globin super-enhancer in vivo
This paper by Hay et al is an excellent read. Often times in genomics we describe phenomena wich we believe have strong biological implications. The discovery and calssifications of super enhancers being one such instance. This paper does a great job in going into a system (mouse) and test the hypothesis that enhancers have emergent qualities. So - if you and all your enhancer buddies are hanging out - do you get stronger,much like Bros lifting in a gym? The answer appears to be… No.
Rather you have an additive effect between enhancers. So, nothing super special.Really calls into question some of the rhetoric that has been deployed around these pieces of the genome.
Other great takeaways:
- Enhancer Assys don’t always line up with genomic prediction. Three reported enhancers appear to have no reporting capability in vivo (granted this might be due to poor reporting assays, or local chromatin envriorments)
- “Super” enhancers appear to offer quite a bit of backup capabilities. Delete one - or two (Figure 4 B/C) and you may not see any change, or any signifigant change in phenotype.
- Looping doesn’t seem to be altered by deletion of the strongest enhancers. For whatever reason topological assocations appear to be tough as nails
- Enhancer Accessible chromatin formation is NOT dependent on other Enhancers. So if you delete the big Kahuna - all the other enhancers form just fine
Key-Figure: 4
Looping
Functional dissection of the Sox9–Kcnj2 locus identifies nonessential and instructive roles of TAD architecture, Despand Et.al (2019)
Loops are a weird and wild place. If you keep up with gene regulation literature at all you’ll know they’re kinda the ‘hot’ ticket item recently for research. With some claiming that loops are an essential, and massive component of gene regulation due to their capactiy of linking up genes with their cognate enhancers, while others argue that they really don’t do as much as we perceive.
Basic model of loop extrusion model (this is going to be displayed as an image because I could write a blog post about the intricacies of how this works)
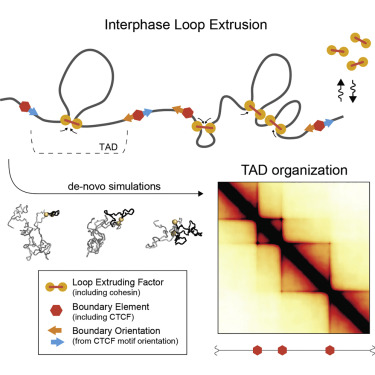
This paper by Despang et al seeks to answer these questions by functionally dissecting, altering and rearaging the TADs around the sox-9 kcnj2 locus. This is an excellent choice considering that both of these genes generate striking phenotypes in mouse development. Paired with this is the advantage that this loci is relatively enhancer rich, allowing the authors to get at one outstanding question in looping-enhancer biology. Mainly - do loops dictate enhancer gene expression, thus altering gene expression?
Main Take Aways:
- CTCF sites and TAD substructure, PAIRED with TAD boundry are what mediate TADs
- Fusing of TADs has no large change on gene expression or phenotype. So… Enhancers appear to be able to find their cognate gene without speicially defined TADs (You’ll just have to take the authors word here that this is happening. Mainly point to phenotype of this last conclustion)
- Inversions don’t alter TADs UNLESS the inversion also contains the TAD boundries (Figure 4b/c). However- kcnj2 DOES have increased contacts with the enhancer elements and does exhibit increased gene expression
- Of the structural re-aragnmetns they caused gain of function mutations. With expression of kcnj2. IN the inversion contining the TAD boundries, kcnj2 actually took up the phenotype of sox9 (wild). This gain of function patter wast still seen just with the fusuion of the TAD boundries, here incDeltaBOR (Figure 5c)
Other great takeaways:
- Fusing TADs doesn’t really seem to matter too much which is intersting.
- The authors point to phase separation as a possible mechanism of how enhancers are still accessing their cognate gene. Will phase separation be the next looping? Time will tell! (yes it will)
- Excellent figures imo
Key Figures: 2-5 tbh. Hugely important paper that’s really worth getting into
lncRNAs
Noncoding Transcription by Alternative RNA Polymerases Dynamically Regulates an Auxin-Driven Chromatin Loop (2014)
This paper is an interesting one because it’s really one of the first paper to find an lncRNA in arabidopsis, delete it, and show a phenotypic effect. This alone makes it a fairly important piece of plant genomics literature in my opinion. Authors on this guy are Ariel et al, with crespi being the senior investigator.
But diving into this paper, this paper seeks to demonstrate the link that an identified lncRNA by the name of APOLO regulates PINOID (PID), a well known gene in root development.
So, to takcle this the authors generated a system to interfere with APOLO using RnaI techniques. Upon knocking down APOLO transcription, PID was knocked down as well, and generated longer roots as a byproduct - Figures 1 B-D.
Next, the authros attempt to demonstrate that the epigenetic landscape of APOLO and PID dynamically change in response to AUXIN treatment. I find this section (Figure 2) fairly poorly explained, and hard to interpret. Their ChIP-qPCR graphs strike me as being nearly indecipherable. That being said they take this study in an intersting direction as they attempt to elucidate how APOLO is being transcribed. They know from mutants like nrpd2a (pol IV subunit) that APOLO was still transcribed meaning it’s most likely transcribed by POL II.
With this paper however, I do have some issues. I think Figure 2 is nearly illegible here. It’s challenging to understand, and the trends to authors point to are rather opaque. Secondly, the mechanism which they draw upon is quite complex, making me wonder well… Is it really that complex? Or have they heaped on a complex mechanism in an attempt to explain this. Added on top of this is recent research in the plant field pointing to the idea that methylation may not be the end all be all of gene silencing, esecially considering that some gene body nethylated genes are heavily transcribed.
Other issues - ChIP-qpcr in 2014 to me seems really… Questionable. Why not do ChIP-seq? There’s no need, and it would have made many of their figures far more convincing in my opinion.
Main Takaways:
- There are lncRNAs in the arabidopsis genome that have phenotypes. I’ve had more than a few poeple ask me if such a thing exist so that’s great to have
- They can be possibly transcribed by different POLs either POL II or POL IV.
- They appear (possibly) to have a capacity to regulat the epigenetic landscape in regards to recruiting PCRC1/2 (not sure I buy this. Other review have said this point is overblow in mammals)
Key Figures: 1
Unlinking an lncRNA from Its Associated cis Element (2016)
I really like this paper overall. I think it does an excellent job of displaying a somewhat novel mechanism of lncRNAs. It’s short, sweet, to the point, and does the exact exerpiments you want them to do. Anyways, to the paper.
This paper by Parakallar et al basically ask the question, “does the lncRNA matter” compared to the sequence underneath it? It does so by looking at the lncRNA Lockd and the gene Cdkn1b. The authors start off by demonstrating that if you delete the lncRNA Lockd using cirps/cas9, the gene product is reduced (Figure 1).
However, if you simply add a polyadenalation signal to the middle of exon 1 of the lncRNA, the lncRNA product is reduce, but the gene product remains almost unchanged. Demonstarting that the lncRNA product itself isn’t important, but the loci is.
It should be noted though, that this doesn’t mean that the lncRNA product itself is TOTALLY useless. Rather, it’s useless to it’s closest gene. The authors didn’t explore the possability that Lockd was acting in Trans in any way. Granted, most of the known lncRNAs (xist mainly) appear to act in a neighborhood esque manner.
Other Takeaways:
- Man, I’m so envious how easy it is to generate knockouts in cell lines. Seems like you’re so unrestrained in some systems (yay plants!)
- Excellent concise review of other mechanisms found in the lncRNA literature, and some other things to brush up on.
Key Figure: 2B
Transcription
Stochastic mRNA Synthesis in Mammalian Cells (2006)
This paper by Raj et al is a pretty sweet study that really began our understanding of the ‘burst’ dynamics of transcription, and man oh man it’s cool. Often times I feel as if the term ‘elegent’ is overused when describing papers, but this one, this one is certainly elegant.
The authors initiated this study by wanting to study transcription in vivo. To do so they develope this awesome system where thye basically create an reporter gene with a ton of binding sites for florescent probes. When expressed and annealed, these probes are able to pinpoint single molecules in the cell.
After demonstrating that this ‘works’ the authors then go on to describe where mRNA localizes. Their big point is that when you see these lit up regions of transcription you generally see more mRNA in the nucleus, and when you don’t see these transcriptionally active points you tend to find more mRNA in the cytoplasm, Figure 2D. They also make a point that in 2D these two cells while having vastly different localization of mRNA also appear to be delinated from the same parent cell, indicating that mRNA variation is not simply due to differences in cell cycle stages.
Next, the authors modify their reporter gene with either one or seven copies of totO, a reporter that you’re able to shut off in the prescence of Doxycycline. Doxycline minimizes the required holodimer tTA required to drive transcription. Bascially they found that increasing the amount of doxycline reduces the amont of mRNA of the gene, but the NOISE, here defined as (standard deviation/mean ) did NOT change. They chock this up to being reflective of the thing that really matters is the activation state of the gene itself.
Then go on to show that increasing the number of TF factors that bind OR increasing the number of transcription factor sites increases the size of the burst. This paper goes on pionting out that it appears genes that genes being turned on is intrinsically random.
Main Takeways:
- Transcription happens in burst, the size of these burst are modulated by thing liks Transcription factor binding and such
- The pinpoint chromatin as a likely cause of which changes genes from being ‘on’ or ‘off’ pointint to the idea that the rate limiting step of transcription is most likely chromatin decondensation.
- Give these results there are most likely two ways in which transcription is able to increase - (ii) increase the rate of transcription when the gene is in the active state, or (iii) decrease the rate of gene inactivation (the opposite behaviors, of course, apply should a cell decide to downregulate a gene’s transcription)
Key Figures: 3